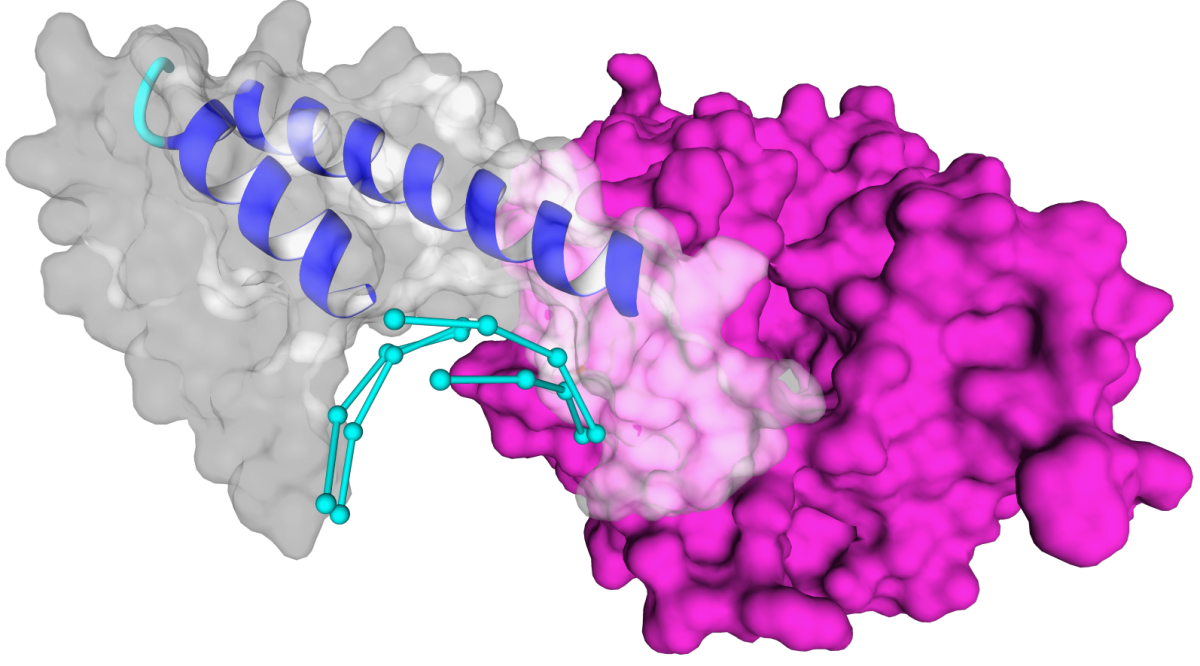
The basic run (using default parameters ) of PADA1 looks like this:
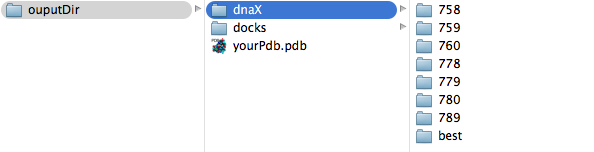
The output of a PADA1 run is composed of two directories: dnaX and docks.

Docked fragments
If you get no dnaX folder it means that you couldn't predict any docked fragment. Inside dnaX folder the predictions are sorted by residue number (and by molecule if mol parameter is set), see example of ouput directory tree below:
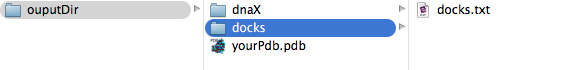
Energy file
Inside docks folder you will find docks.txt with all the information of the docked fragments. The docked fragments are written in pdb format without the input protein to save storage, to visualize them you need to open both, the fragment and the input protein, at a time in any molecular viewer.
The typical ouput for a docked fragment inside docks.txt looks like this:
Molecule: A Residue: DT 504 -0.165
Molecule: A Residue: DC 505 0
Molecule: A Residue: DG 506 0
Molecule: A Residue: DC 507 0
Molecule: B Residue: DG 527 -0.536
Molecule: B Residue: DC 528 -0.251
Molecule: B Residue: DG 529 0
Molecule: B Residue: DA 530 0
The first information line contain the fields: inputPdb, beginResidue of the scanPeptide, database peptide id, database dna fragment id, forward strand sequence of the dna fragment, reverse strand sequence of the dna fragment, force field energy, number of contacts. The following lines describes the information about the nucleotides and the contribution for each of the to the free energy of binding calculated with PADA1 Boltzmann device. As the parameter residue is not set the algorithm will run a full scan over the input protein using overlaping sliding windows (scan peptides) along the protein. If you set residue parameter you can explore specific regions and even paralelize the run. An example on parallel runs can be extrapolated from the following lines:
Exhaustive Mode
If you get no results using the defaults parameters you can explore more posibilities (more time consuming) for instance by increasing the pep-mismatches value:
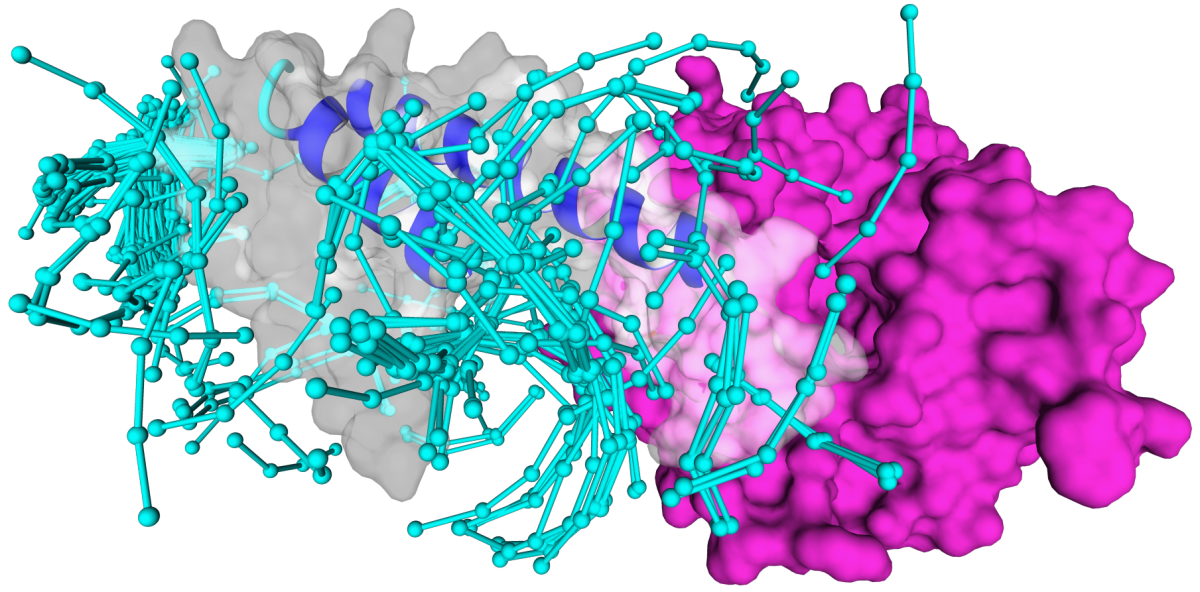
If you get a highly populated fragment cloud, you can reduce off targeting by setting the parameter min-contacts (3-4 is a reasonable value) and rank the results by energy: